Or: How I Learned to Stop Averaging Coordinates and Embrace the Fused Gromov-Wasserstein Barycenter
TL;DR: Predicting chemical properties accurately usually forces you to choose between fast 2D molecular graphs or computationally heavy 3D structures (conformers). We built CONAN-a framework that merges 2D graphs with multiple 3D molecular structures using an -invariant , differentiable Fused Gromov-Wasserstein Barycenter solver. It sets a new state-of-the-art across MoleculeNet benchmarks while using significantly fewer conformers.
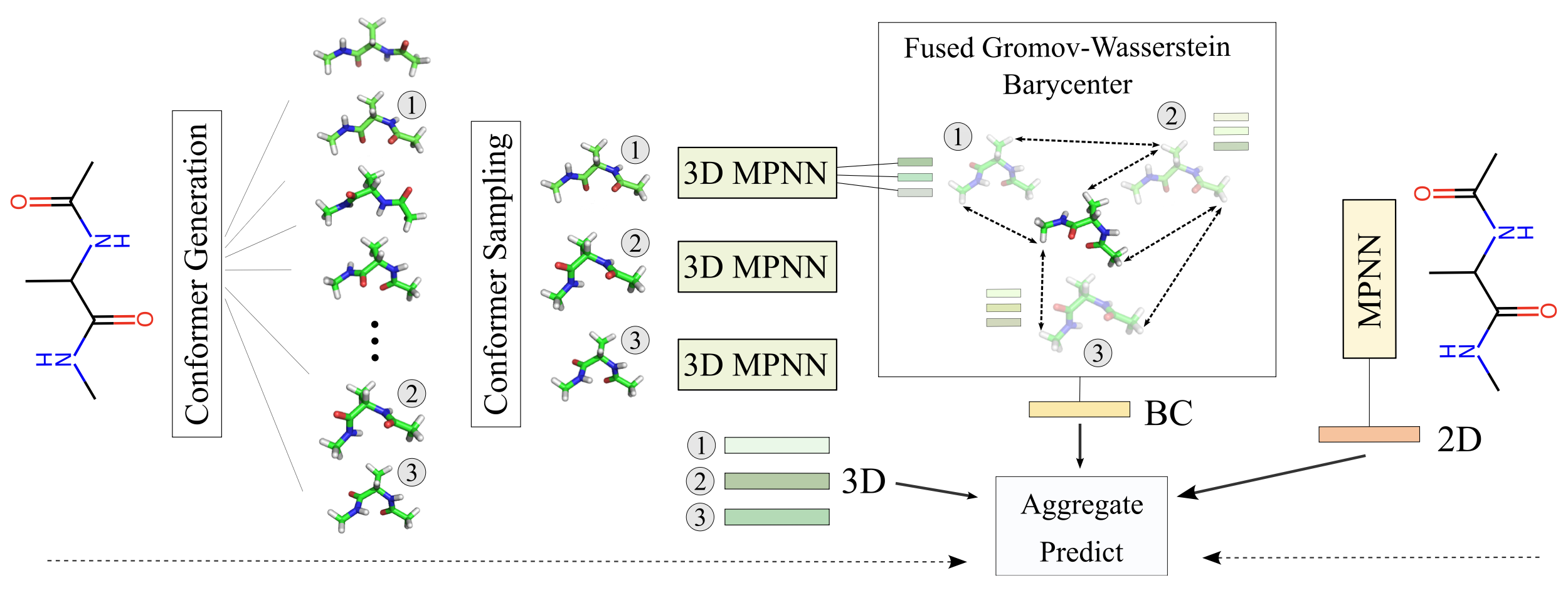
This all started because looking at molecules in isolation can be incredibly limiting. Standard machine learning methods for property prediction tend to look either at a flat 2D schematic or a single, rigid 3D snapshot. But in nature, a molecule changes shapes constantly , and properties like solubility or binding affinity depend on an entire ensemble of these 3D conformations.
Our solution? We proposed CONAN (CONformer Aggregation Networks). Instead of relying on a single structural geometry , it pools information across several 3D variations alongside traditional 2D representations.
The Evolution of a Pooling Hack
If you try to capture the essence of multiple 3D conformers, the immediate temptation is to do standard arithmetic mean pooling on the atom coordinates. It works, right? Well, sort of… until you run into simple geometric transformations.
Consider two identical conformers where one is just a 180-degree rotation of the other. If you blindly average their raw Cartesian coordinates, the atoms collapse mathematically into the exact same central position. You end up generating an unphysical, mashed-up ghost structure that completely breaks your machine learning model.
That’s when we fell down the optimal transport rabbit hole. We needed an aggregation mechanism that respects physical reality , permutation symmetries , and actions of the Euclidean group -meaning it stays perfectly invariant to translation, rotation, and inversion.
Plot Twist: Geometry-Aware Barycenters
To avoid collapsing coordinates, we treat our 3D spatial arrangements and atom embeddings as structured objects. Rather than averaging points in space, we solve for a Fused Gromov-Wasserstein (FGW) Barycenter.
Think of the barycenter as a structure-preserving pooling operation. It acts as a geometric mean that simultaneously aligns the structural distance matrices (how far apart atoms are from each other) and feature distances (latent atomic properties) across all sample shapes.
The result? The mathematical architecture ensures perfect -invariance and handles conformers seamlessly as an unordered set. Even better, our theoretical analysis proves that this empirical barycenter converges to the true target structure at a rapid rate of , where is the number of conformers. This means you don’t need hundreds of expensive 3D structures; a modest handful (like 5 or 10) gives an incredibly clean approximation.
The Problem: Computational Bottlenecks
Here’s what happens when you try to use exact Fused Gromov-Wasserstein distances inside a heavy deep learning pipeline:
-
You hit massive scalability challenges.
-
Standard optimization techniques rely on Conditional Gradient algorithms that run slow, single-threaded operations.
-
You can’t easily build clean auto-differentiation computation graphs over classic network flow solvers.
Without fixing this, training a molecular model on parallel hardware becomes a pipe dream.
The Solution: Entropic Scaling on GPUs
To make this practical for real-world chemistry and drug discovery, we introduced two crucial optimizations:
1. Distance Geometry Sampling
Instead of spending massive computing power calculating high-precision molecular physics upfront, we use efficient distance geometry-based sampling through RDKit. We convert rough boundary bounds and constraints directly into fast 3D coordinate drafts.
2. The Entropic Solver
We apply an entropic relaxation term () to the transport problem. This turns a rigid mathematical hurdle into a smooth objective that can be solved via vectorized Sinkhorn projections.
# Conceptual loop for Entropic FGW Barycenter updates
for s in range(K):
# Solve entropic alignments using stabilized Log-Sum-Exp operations
cross_couplings[s] = solve_entropic_ot(barycenter_graph, input_conformers[s])
# Update structural matrices smoothly without coordinate collapse
barycenter_A = update_structure(cross_couplings, input_structures)
barycenter_H = update_features(cross_couplings, input_features)
By leveraging stabilized log-sum-exp (LSE) operators , our training pipeline scales linearly with the number of conformers () , while previous methods like FGW-Mixup scale exponentially. We can pass gradients backward right through the solver iterations across multiple GPUs in parallel.
Testing Our Implementation
We benchmarked CONAN-FGW against state-of-the-art molecular regression tasks from MoleculeNet.
Molecular Property Prediction (RMSE ↓)
| Model | Lipo | ESOL | FreeSolv | BACE |
|---|---|---|---|---|
| 2D-GAT | 1.387 | 2.288 | 8.564 | 1.844 |
| MolFormer (Pre-trained on 1.1B compounds) | 0.700 | 0.880 | 2.342 | 1.047 |
| UniMol (Pre-trained on 209M structures) | 0.603 | 0.788 | 1.480 | - |
| CONAN-FGW (Ours, no pre-training) | 0.487 | 0.529 | 1.068 | 0.549 |
Our framework consistently out-performed massive, heavily pre-trained chemical foundation models. It also showed significant margins on complex 3D SARS-CoV classification benchmarks while using up to 40x fewer conformers than traditional pipeline alternatives.
The Bottom Line
Effectively mapping molecular structures requires:
-
Moving past flat 2D spaces into multi-conformation 3D structures.
-
Avoiding raw coordinate averaging to prevent unphysical structures.
-
Utilizing Fused Gromov-Wasserstein Barycenters for optimal shape-pooling.
-
Applying entropic regularized Sinkhorn iterations to keep training highly scalable and parallelizable on modern hardware.
Building deep models for drug discovery is hard mode. But with the right geometric tools, you can let optimal transport do the heavy lifting for you.
@InProceedings{pmlr-v235-nguyen24g,
title = {Structure-Aware E(3)-Invariant Molecular Conformer Aggregation Networks},
author = {Nguyen, Duy Minh Ho and Lukashina, Nina and Nguyen, Tai and Le, An Thai and Nguyen, Trungtin and Ho, Nhat and Peters, Jan and Sonntag, Daniel and Zaverkin, Viktor and Niepert, Mathias},
booktitle = {Proceedings of the 41st International Conference on Machine Learning},
pages = {37736--37760},
year = {2024},
editor = {Salakhutdinov, Ruslan and Kolter, Zico and Heller, Katherine and Weller, Adrian and Oliver, Nuria and Scarlett, Jonathan and Berkenkamp, Felix},
volume = {235},
series = {Proceedings of Machine Learning Research},
month = {21--27 Jul},
publisher = {PMLR},
pdf = {https://raw.githubusercontent.com/mlresearch/v235/main/assets/nguyen24g/nguyen24g.pdf},
url = {https://proceedings.mlr.press/v235/nguyen24g.html}
}